Plant Genome Research
In the last decade, the sequencing and annotation of complete plant genomes has helped us understand plant function and evolution, as well as how to alter economically important traits. Efforts in many disparate disciplines are required to generate reference genomes. The work starts with laboratory scientists, who generate the raw sequence data. Next, computational biologists and bioinformaticians, such as in Ware lab, kick off a series of steps to interpret the raw data. The process of interpretation involves the assembly of raw sequence reads into overlapping segments (“contigs”), which are combined to create a scaffold. This scaffold, in turn, discerns the position, relative order, and orientation of contigs within the chromosomes. The next step is annotation, the discovery and description of genes and other functional elements, and homologies (evolutionary relationships) with other genomes. This information must be faithfully communicated and visualized in web-based platforms, such as Gramene.
All of these activities are rapidly evolving with the advent of new sequencing technologies, algorithms, and data-handling requirements. For example, high-depth and low-cost sequencing of RNA transcripts is providing a vast stream of new evidence that informs genome annotation; this in turn has spurred the development of new software for modeling and performing genome annotation. Low-cost sequencing has also made it possible to ask whole new classes of questions, moving beyond the generation of single references for individual species and supporting the development of multi-species representation as a “pan-genome”. Ongoing projects within the maize, rice, and Arabidopsis research communities are now sequencing hundreds or thousands of genotypic backgrounds, chosen from carefully constructed populations, wild populations, and breeding germplasms in each species. Information about genetic variation is helping scientists to understand the genetic basis of phenotypic traits and the origins of domesticated crop species. New technologies are also driving research in epigenetics, the study of heritable variation not caused by changes in the underlying genome sequence. Epigenetic mechanisms include modification of DNA by methylation and various forms of histone modification that can cause changes in gene expression and other phenotypes. Both types of modifications can be studied using new sequencing technologies and analytical methods.
Updating the Maize B73 Reference Genome and Annotation using Long Sequence Read Technologies.
Maize is an important feed and fuel crop as well as model system in developmental genetics. A complete and accurate reference genome is imperative for sustained progress in understanding the genetic basis of trait variation and crop improvement in maize. The Ware laboratory has played a key role in development and stewardship of the maize reference genome since the inception of the maize genome project almost a decade ago. Although the current B73 reference sequence has seen incremental improvements, many gaps and mis-assemblies remain due to technical limitations in sequence technologies relative to the complexity of the maize genome. To remedy this, we are employing PacBio long read sequencing technologies to build the next generation maize reference genome. A total of ~65X coverage of B73 genome long reads were generated. The N50 of the reads in this data set is nearly 15kb, with 43.5% of reads longer than 10kb. To construct a de novo assembly, we have employed the PBcR-MHAP correction and assembly pipeline. After optimization, this method yielded an assembly of 3,303 contigs with a total length of 2.10Gb. The N50 and average size of the contigs reached 1Mb and 634Kb respectively.
We are currently working with optical maps to further evaluate these assemblies and to aid with construction of pseudomolecules. We are also developing an efficient maize gene annotation pipeline to support the release of this new version of maize reference genome. We are also using the same single-molecule sequencing technology to investigate the maize transcriptome.
A Sorghum Mutant Resource as an Efficient Platform for Gene Discovery in Grasses
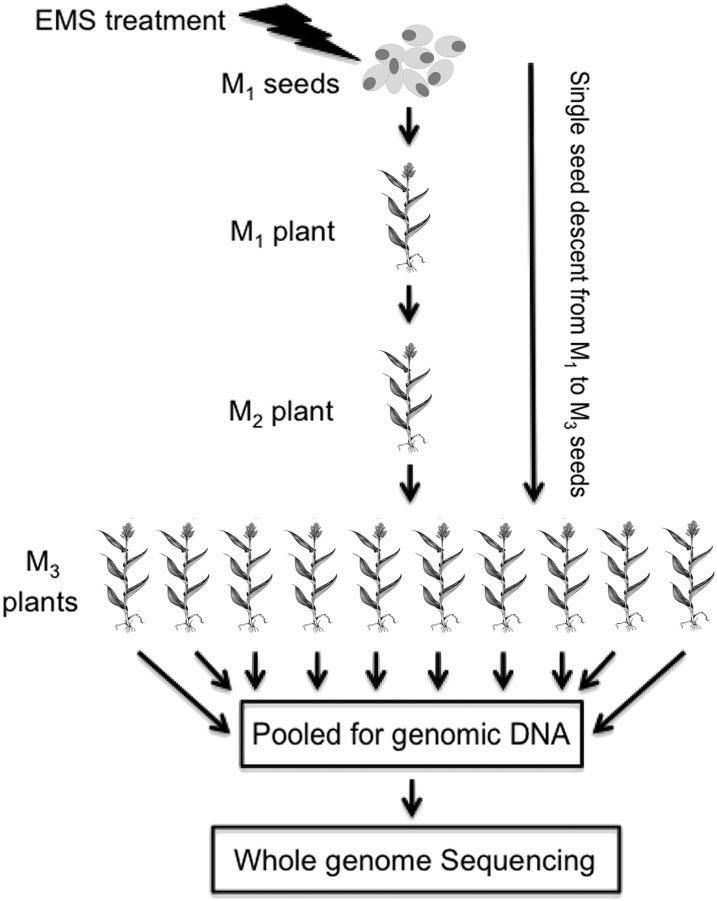
Sorghum (Sorghum bicolor) is a versatile C4 crop and a model for research in family Poaceae. High-quality genome sequence is available for the elite inbred line BTx623, but functional validation of genes remains challenging due to the limited genomic and germplasm resources available for comprehensive analysis of induced mutations. In this study, we generated 6400 pedigreed M4 mutant pools from EMS-mutagenized BTx623 seeds through single-seed descent. Whole-genome sequencing of 256 phenotyped mutant lines revealed >1.8 million canonical EMS-induced mutations, affecting >95% of genes in the sorghum genome. The vast majority (97.5%) of the induced mutations were distinct from natural variations. To demonstrate the utility of the sequenced sorghum mutant resource, we performed reverse genetics to identify eight genes potentially affecting drought tolerance, three of which had allelic mutations and two of which exhibited exact cosegregation with the phenotype of interest. Our results establish that a large-scale resource of sequenced pedigreed mutants provides an efficient platform for functional validation of genes in sorghum, thereby accelerating sorghum breeding. Moreover, findings made in sorghum could be readily translated to other members of the Poaceae via integrated genomics approaches
Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing.
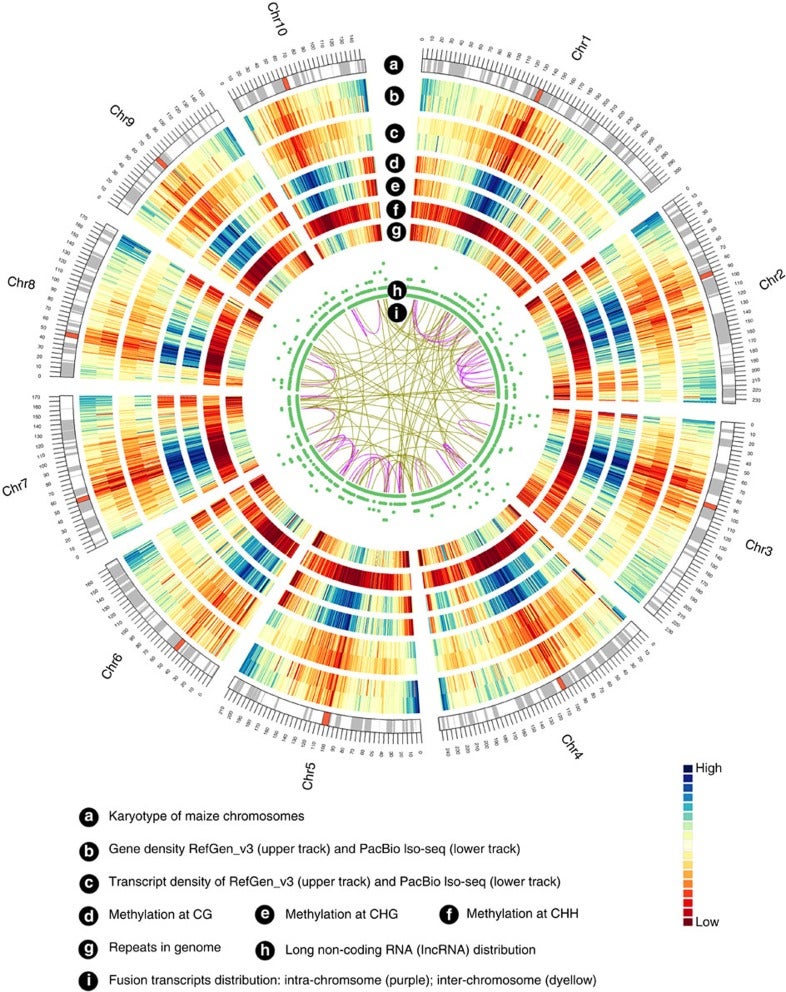
Zea mays is an important genetic model for elucidating transcriptional networks. Uncertainties about the complete structure of mRNA transcripts limit the progress of research in this system. Here, using single-molecule sequencing technology, we produce 111,151 transcripts from 6 tissues capturing ∼70% of the genes annotated in maize RefGen_v3 genome. A large proportion of transcripts (57%) represent novel, sometimes tissue-specific, isoforms of known genes and 3% correspond to novel gene loci. In other cases, the identified transcripts have improved existing gene models. Averaging across all six tissues, 90% of the splice junctions are supported by short reads from matched tissues. In addition, we identified a large number of novel long non-coding RNAs and fusion transcripts and found that DNA methylation plays an important role in generating various isoforms. Our results show that characterization of the maize B73 transcriptome is far from complete, and that maize gene expression is more complex than previously thought.
Gramene: Comparative Genomic Resource for Plants
The Gramene project provides online reference resources for plant genomes and curated pathways to aid functional genomics research in crops and model plant species. Our website, www.gramene.org, facilitates studies of gene function by combining plant genome and pathway annotation with experimental data and cross-species comparisons. In other words, the data and tools in Gramene enable plant researchers to use knowledge about gene function in one species to predict gene function in other species. This ability should advance our fundamental understanding of plant physiology, including economically important traits such as hybrid vigor, grain development, seed dormancy, flowering time, drought tolerance, and resistance to diseases. Last year we significantly streamlined the user interface and backend functions.
For the first time, Gramene processed EMS-mutation data for inclusion in its genome browser. The Gramene team develops comparative genomics databases in collaboration with the Ensembl Plants project at the European Bioinformatics Institute (EBI). Perhaps the most significant achievement from this collaboration was the new bread wheat chromosome-scale pseudomolecules assembly using existing IWGSC CSS contigs and incorporating POPSEQ data from the Leibniz Institute of Plant Genetics and Crop Plant Research (IPK) and processed by The Genome Analysis Centre (TGAC).
Gramene is building integrated data stores from these primary data sources to complement the REST APIs offered by Ensembl and Reactome. Lightweight node.js web servers provide HTTP access to the integrated data and auxiliary structured annotations stored in MongoDB and Solr document collections. Unified access to these services is provided through http://data.gramene.org, which is built using swagger middleware. The Gramene search interface (http://search.gramene.org; Figure 3) differs from most other search interfaces in that it guides the user to select filters based on gene identifiers, names, and descriptions as well as structured annotations such as InterPro domains, Gene Ontology terms, and Plant Reactome pathways


Plant Systems Biology
Exploring Arabidopsis Gene Regulatory Networks
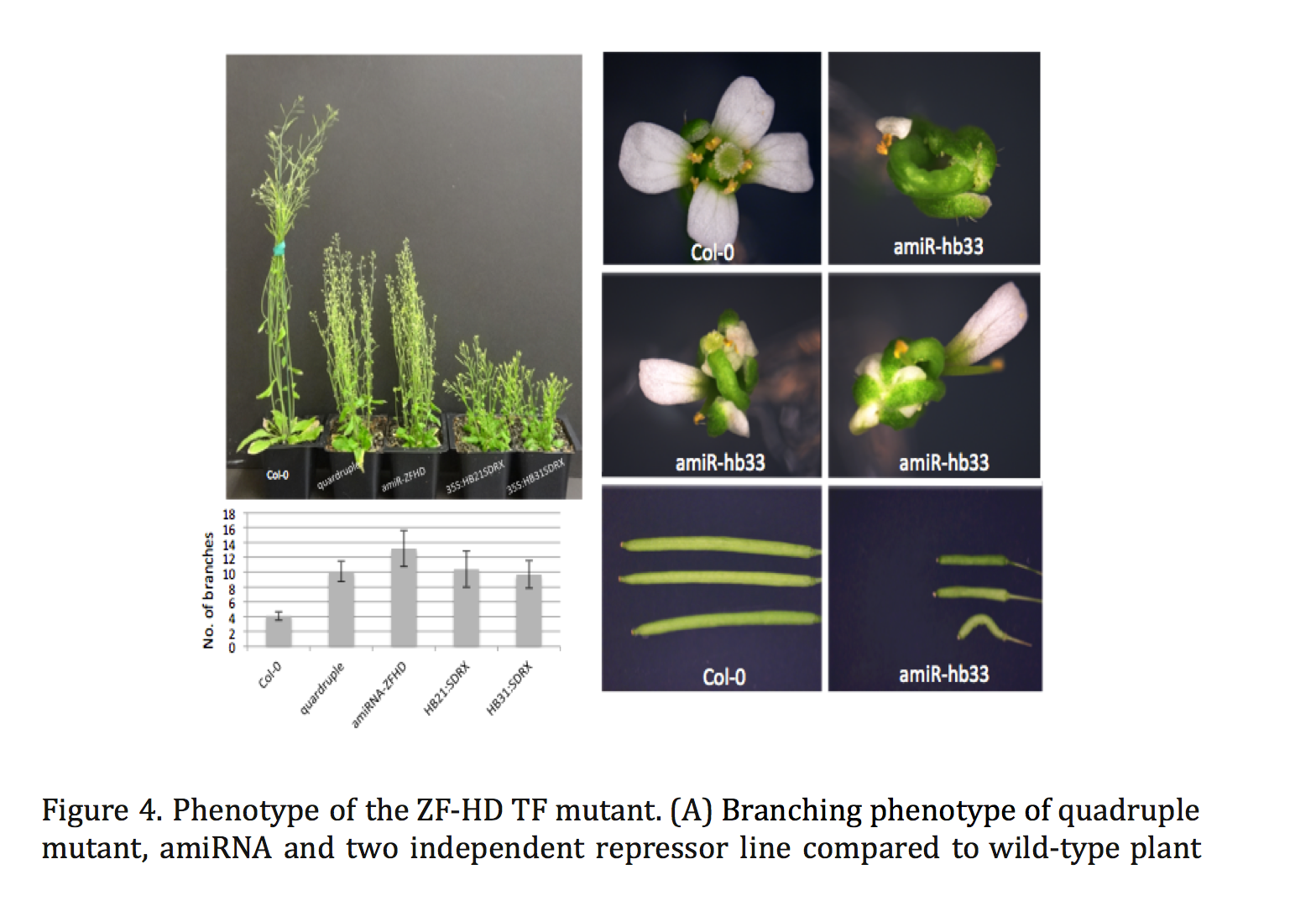
Plant miRNAs play important roles in regulating plant development and stress responses by post-transcriptionally repressing the expression of their target genes. To identify upstream regulators of miRNA expression, we generated the Arabidopsis miRNA Gene Regulatory Network (ARMIG) using a yeast one-hybrid (Y1H) approach. Using a nearly complete root transcription factor (TF) library, we screened 180 miRNA promoters, their targets, and TFs that are highly connected within the network, and obtained 5376 Protein-DNA interactions (PDIs). ARMISs are characterized by highly connected genes as “hubs” in the network. The ZF-HD TFs were identified as hub genes within the miRNA network
Combining multiple loss of function mutants within a single Arabidopsis line we observed several phenotypes, including altered flower structures and an increased vegetative branching. In addition, we performed transcriptome profiling of ZF-HD TF mutants in order to characterize differentially expressed genes. The gene-expression profiles and phenotypic properties of the mutants suggested a role for ZF-HD TFs as regulators of developmental transitions. Moreover, the results of this work demonstrate that the miRNA GRN can be applied more generally, beyond the root system.
Developmental Networks Controlling Inflorescence Architecture in Grasses
The goal of this work is to integrate genetics and genomics data sets to find molecular networks that influence the morphology (architecture) of grass inflorescences (flowers). Because inflorescences bear the fruits and grains that we eat, the genetic and regulatory factors that govern their formation are clearly relevant to important agronomic traits such as grain yield and harvesting ability. Our work in maize focuses on inflorescence primordia sampled during key developmental transitions and in perturbed genetic backgrounds. The latter includes loss-of-function mutants in three important regulators of the RAMOSA (RA) pathway, which controls stem cell fate decisions and ultimately the decision to branch. We established a robust system for investigating the networks that modulate branching, including characterization of precise timing of developmental events and the associated spatiotemporal changes in gene expression. We integrated genome-wide mRNA-seq data to resolve co-expression networks during key stages of maize inflorescence development, and we are currently working to expand these networks by incorporating additional data sets, including genome-wide transcription factor (TF) occupancy profiles and cis-regulatory information.
Cyberinfrastructure Projects
CyVerse (formerly The iPlant Collaborative)
Advances in biological research technology have enabled scientists to amass unprecedented amounts of data, and many researchers find themselves drowning in this sea of data. Foreseeing this major bottleneck in biological research, the U.S. National Science Foundation (NSF) established the iPlant Collaborative (http://iplantcollaborative.org) in 2008 to develop cyberinfrastructure for life sciences research and democratize access to U.S. supercomputing capabilities.
Having completed its first 7-year grant, the CyVerse Collaborative has made extensive progress toward meeting these goals. Work in the last year culminated in the development of multiple platforms that are now being used by the research and educational communities. Staff at CSHL contributed directly to some of these cyberinfrastructure platforms, or built upon existing platforms to provide scientists and educators with ready access to needed software and analysis tools. Within the Ware lab, these platforms include the CyVerse Data Store, Discovery Environment (DE), and Atmosphere. In addition, Ware lab has successfully built the first federated CyVerse system at CSHL that supports a dozen HPC apps running on a local cluster and storage system.
A major mission of CyVerse is to promote adoption of the cyberinfrastructure through training workshops and outreach at academic institutions and scientific meetings. In 2015, members of the Ware lab participated as instructors in several “Big Data” workshops focused on transcriptomics and population genomics using the DE and Atmosphere platforms.




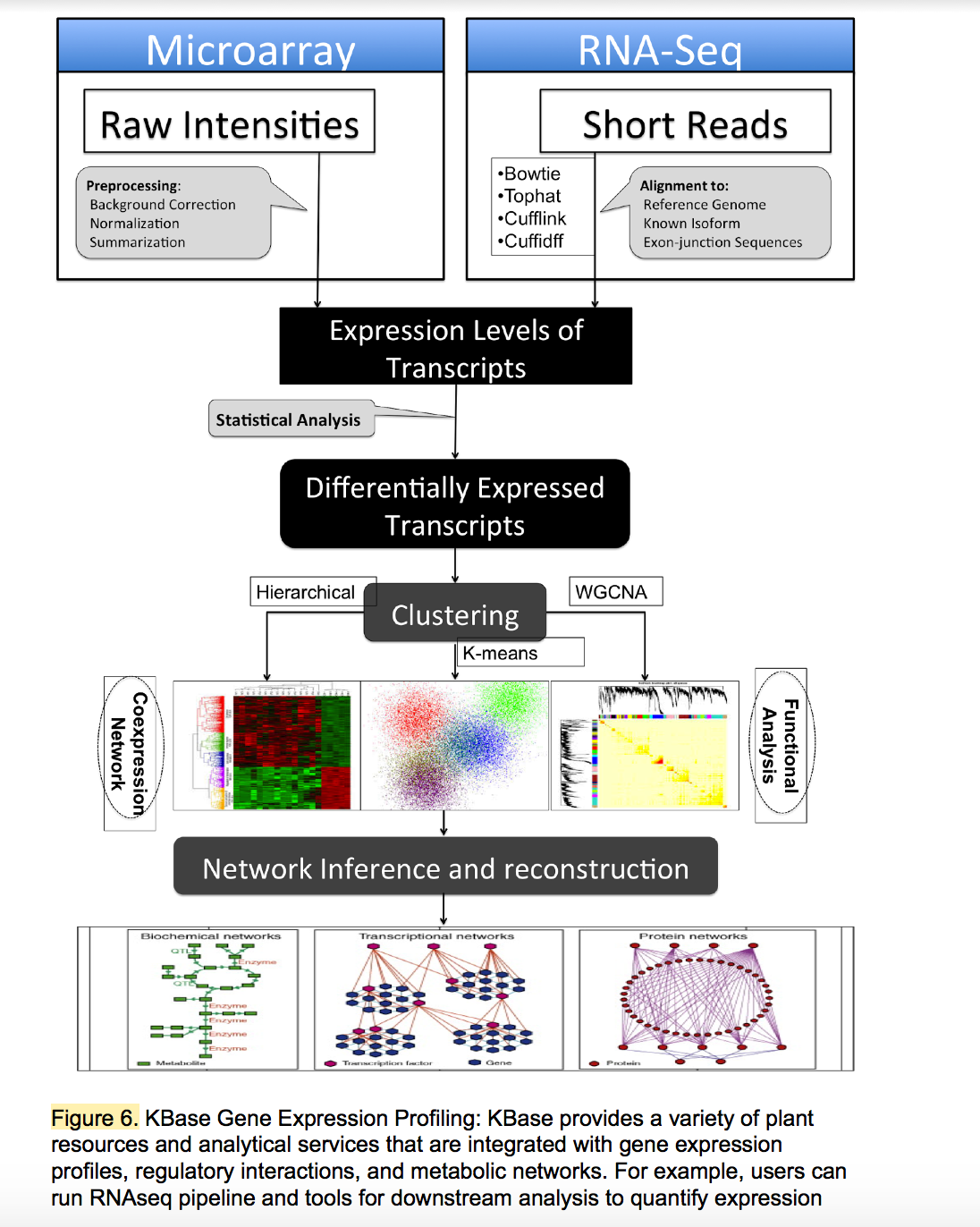
KBase: Department of Energy Systems Biology Knowledgebase
The Systems Biology Knowledgebase (KBase, www.kbase.us) has two primary goals. The scientific goal is to produce predictive models, reference datasets, and analytical tools, and to demonstrate their utility in DOE biological research relating to bioenergy, carbon cycle, and the study of subsurface microbial communities. The operational goal is to create the integrated software and hardware infrastructure needed to support the creation, maintenance, and use of predictive models and methods for the study of microbes, microbial communities, and plants.
KBase’s computational infrastructure is supported by a distributed, high-performance, cloud-based system that includes more than 3 petabytes of storage, over 12,000 cores for data processing, and 90 GBit/s bandwidth over DOE’s ESnet. The KBase data model supports more than 900 data types, including sequence reads, assemblies, genomes, annotations, expression data, quantitative phenotype data, and metagenomic profiles. The KBase data repository has integrated various datasets from public resources for thousands of microbes, hundreds of microbial communities, and tens of plants.
In addition, KBase supports more than 1,000 analysis and access functions grouped into more than 30 integrated service modules, including sequence assembly, variation analysis, genome annotation, expression analysis, and various types of modeling (e.g. metabolic, regulatory, and flux balance analysis [FBA]). In order to capture complex, multi-step analyses, users can build persistent and provenance-based workflows that can capture complex, multi-step analyses and mobilize rich annotations, visualization widgets, reusable workflows, and custom scripts. This capability empowers users to create science that is transparent, reproducible and reusable.